
孕知因-多項遺傳疾病帶因篩檢
文章目錄
• 單基因遺傳疾病風險高過唐氏症
• 夫妻雙方身體無恙 寶寶竟有遺傳疾病?
• 台灣常見單基因隱性遺傳疾病有哪些?
• 病童沒有家族史!檢查才知爸媽皆帶因者
• 揪出隱藏疾病!孕知因帶因篩檢一生只需測一次
• 國際醫學會共同建議 這些人需要帶因者篩檢
• 婦產醫師推薦孕知因
• 孕前、孕期守護3重奏 帶給寶寶無憂的未來
• 孕知因-多疾病帶因篩檢 檢測流程
• 為什麼帶因檢測要選訊聯基因
• 遺傳疾病帶因篩檢常見問答
• 遺傳疾病案例,聽聽醫師怎麼說
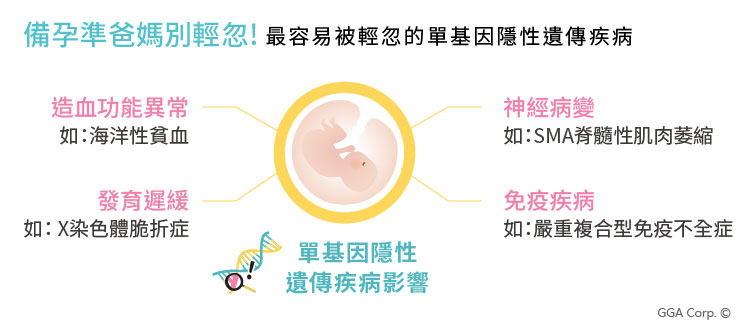
單基因遺傳疾病風險高過唐氏症
根據世界衛生組織統計,全球有超過 10,000 種單基因遺傳疾病,其綜合發生率竟高達1%【參考資料1】,為唐氏症發生率的8倍 (一般文獻統計唐氏症平均發生率為0.125%)。更可怕的事情是,高達80%的單基因遺傳疾病的孩童回溯其雙親都沒有任何家族病史【參考資料2】!
然而,但對比其高發生率,每100位患病孩童中,卻僅有5位有藥物與治療方法【參考資料2】,也因此,多數單基因相關的疾病,往往導致孩子一出生即面臨身體結構缺陷、代謝異常,嚴重者甚至是死亡,影響寶貝的健康甚鉅!
為避免將潛在的未知疾病帶因遺傳給孩子,建議計畫懷孕的夫妻,於孕前進行【孕知因-多疾病帶因篩檢】,以確認是否有潛在的遺傳疾病風險,給予小孩健康的未來。


參考資料:
1. World Health Organization. [Accessed August 19, 2016];Genes and human disease. http://www.who.int/genomics/public/geneticdiseases/en/index2.html
2. National Policy for Treatment of Rare Diseases [Internet]. Misintry of health & family Welfare. [cited 2021 Nov 29]
3. The American Journal of Human Genetics 91, 1022–1032, 2012.



夫妻雙方身體無恙 寶寶竟有遺傳疾病?
-
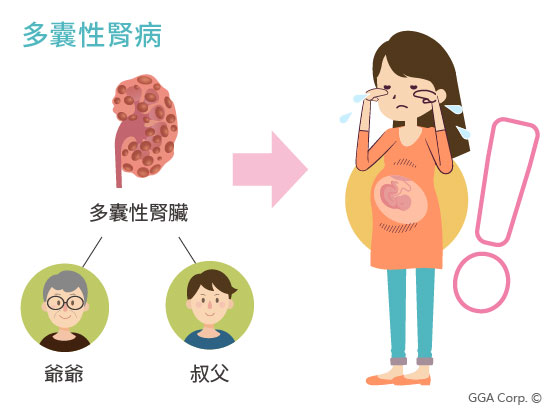
隱性遺傳疾病案例一:多囊性腎病
高雄有位A小姐自從生下大女兒後,遲遲無法懷上第二胎,好不容易透過試管療程成功懷孕,就在孕期28週時進行高層次超音波檢查,發現胎兒的腎臟長滿許多小水泡,進一步確診為遺傳性疾病「多囊性腎病」。
由於一家大小向來都沒有腎臟方面的相關疾病,也從來不覺得自己跟丈夫是帶因者,直到這胎懷了兒子,遺傳到家族多囊性腎病的症狀,她才驚覺和老公竟然同樣都是帶因者。
-
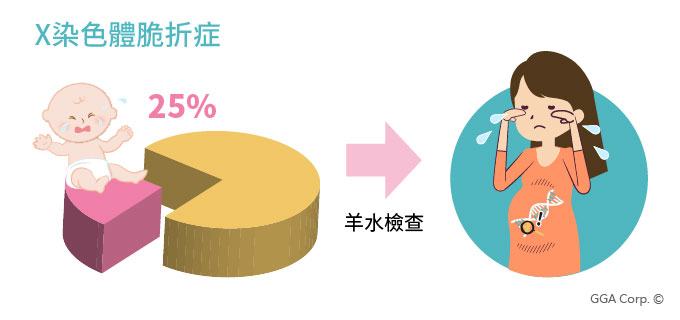
隱性遺傳疾病案例二:X染色體脆折症
台南有對姊妹,姐姐當初懷孕時,見周遭的友人沒做任何檢查,也能生下健康的孩子,便決定只做公費產檢。直到孩子一兩歲時發展緩慢,她才驚覺孩子可能有智能障礙,帶去檢查後,才知道自己是X染色體脆折症的帶因者。害怕的姐姐趕緊告知已懷孕三個月的妹妹,果然妹妹同樣也是帶因者,胎兒經過羊水檢查確認也患有此病,便在妊娠5個月時決定引產。
姊妹倆從來沒想過自己會是疾病帶因者,明明家裡人都沒有任何症狀。她們自責地認為,如果自己能在懷孕前就先做篩檢,也許能避免這樣的遺憾…。
(以上均為真實案例)
台灣常見單基因隱性遺傳疾病有哪些?
以上這樣的案例,在台灣正不斷上演。幸運的是,隨著基因技術的發展,透過次世代定序技術,一次檢測即可評估多項單基因隱性遺傳疾病的【孕知因-多疾病帶因篩檢】,幫助夫妻提前確認彼此是否同為遺傳疾病的帶因者,降低生下罕病兒的機率。
以下這些台灣常見的高帶因率的隱性遺傳疾病,現在都可以透過多疾病帶因者篩檢一次檢出:
| 疾病名稱 |
帶因率 |
Alpha-海洋性貧血(Alpha-Thalassemia) |
1/20 |
Beta-海洋性貧血(Beta-Thalassemia) |
1/100 |
遺傳性耳聾(Hereditary hearing loss) |
1/25 |
裘馨氏肌肉萎縮症(Duchenne Muscular Dystrophy) |
1/30 |
脊髓性肌肉萎縮症(Spinal Muscular Atrophy) |
1/40 |
先天性腎上腺增生症-21-羥化酶缺失症 |
1/61 |
龐貝氏症(Pompe Disease) |
1/70 |
苯酮尿症(Phenylketonuria) |
1/85 |
威爾森氏症(Wilson’s Disease) |
1/85 |
參考資料:台灣罕見疾病基金會
80%病童沒有家族史!檢查才知爸媽皆帶因者
-
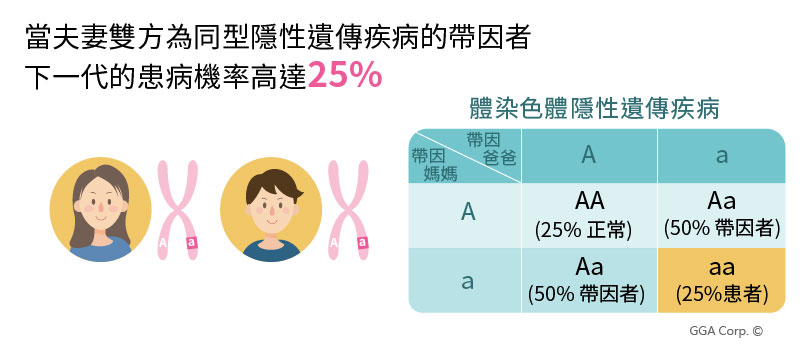
人體共有23對染色體,每一對均有2條染色體分別來自爸爸和媽媽。
-
帶因指一對染色體中其中一條染色體上帶有與隱性遺傳疾病相關的基因突變,且因為只有一條染色體有基因變異,另一條正常,所以帶因者通常健康與外觀上都無異常。
-
然而,當爸爸和媽媽都是同型隱性遺傳疾病的帶因者,下一代的患病機率高達25%
-
據統計,80%的患病孩童回溯其雙親,均沒有任何的家族史,因此除發生率高以外,更棘手的是單基因隱性遺傳疾病的風險多數不易由一般產檢察覺(唐氏症篩檢、非侵入性檢查NIPT、羊膜穿刺、超音波檢查),往往等到寶貝出生後發現患病,父母雙方才驚覺自身帶有的危險因子。
-
所以容易導致爸媽健康,卻生下患病小孩的情形,對於任何爸爸媽媽來說是措手不及的情況。而這些遺傳疾病,往往需要終身的治療與照護,甚至會威脅到小孩的生命,對任何爸媽來說都是沉重的負擔。
因此,隱性遺傳疾病或許並沒有離我們這麼遙遠,它其實就在暗中伺機威脅寶寶的健康!
一生測一次 孕知因-多疾病帶因篩檢幫助妳
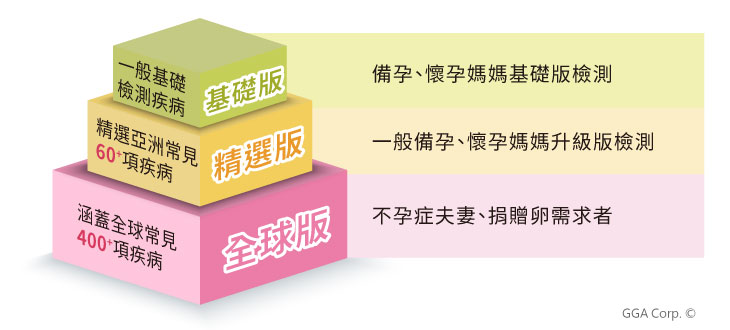
「孕知因-多疾病帶因篩檢」可幫助計畫懷孕或已懷孕的夫婦,提前了解自己的基因狀況,以及寶寶患病的風險,才能盡早與醫師諮詢合適的孕產前檢測,或是討論後續治療的方法。從基因了解胎兒的患病風險,及早檢測,及早準備,在現今少子化的社會中,期望幫助所有家庭都能放心的孕育下一代。
我們依據每對夫妻的需求,精選檢測遺傳疾病,並設計了不同方案,如想知道詳細的疾病列表,可直接點擊下面連結:

國際醫學會共同建議 這些人需要帶因者篩檢
根據ACMG(美國醫學遺傳學暨基因體學學會)、ACOG (美國婦產科學會)以及NSGC (美國遺傳諮詢學會)等學會均提出有生育規劃的族群,皆可進行此項檢測,尤其是以下的族群:
-
想了解自己是否為某隱性遺傳疾病帶因者
-
有遺傳疾病家族史
-
血緣關係較近的夫婦
-
計畫懷孕或已懷孕者
-
精卵捐贈者
參考資料-Obstet Gynecol 2015;125:653–62
Obstet Gynecol. 2017 Mar;129(3):595-596
婦產醫師推薦孕知因

孕知因寵愛孕媽咪 專屬加值服務「滿分守護」
每個想懷孕的媽媽,總是將自己的愛,全心全意放在寶寶身上,反而忘了體貼自己,忘了健康的自己才能陪伴寶寶成長。
寵愛孕媽咪,我們推出【孕知因精選版】 + 【癌症風險基因檢測】+【康知因-成人健康風險檢測】 = 滿分守護 預約婚後幸福

孕知因精選版多疾病帶因篩檢 |
康知因成人健檢基因篩檢 |
癌症風險基因篩檢 |
|
精選60+亞洲與多種族常見遺傳性疾病,降低遺傳疾病對寶寶健康的威脅 包括以下常見疾病:
|
潛藏在DNA的健康因子,透過基因檢測為您把關 檢測30+基因位點,包含以下疾病:
|
幫助您瞭解遺傳性癌症的風險,制定個人化的健康計劃 檢測20+常見遺傳性癌症基因位點,包含以下常見癌症:
|
孕前、孕期守護3重奏 帶給寶寶無憂的未來
|
|
第1重 | 第2重 | 第3重 |
| 項目 |
孕知因 |
SNP羊水晶片 |
預知因寶寶檢測 |
| 時間 |
懷孕前~懷孕14週 |
懷孕16週以上 |
與SNP HD羊水晶片 |
| 方式 |
抽血 |
羊膜穿刺 |
|
| 檢測 內容 |
單基因遺傳疾病 |
染色體異常疾病 |
全方位生活照護基因資訊 |
孕知因-多疾病帶因篩檢 檢測流程
|
Step 1 |
Step 2 |
Step 3 |
Step 4 |
|
|
|
|
|
|
選擇孕知因疾病篩檢 |
提供血液檢體 |
獲得檢測報告 (六週) |
與醫生討論生育規劃 |




為什麼帶因檢測要選訊聯基因
-
超資深的專業遺傳諮詢團隊
-
擁有認證實驗室,且操作經驗超過20年
-
超過150萬次臨床檢測的服務經驗
-
我們擁有專業團隊配送檢體,嚴格掌控檢體的品質
-
我們擁有一條龍的檢測服務,若您夫妻雙方都有相同帶因,且有生育的需求,我們可銜接UPGD超快速胚胎著床前檢測服務,且擁有全台最大的臍帶血資料庫比對

如果您想更進一步了解孕知因-多疾病篩檢篩檢的費用與疾病內容,可以直接☛聯絡我們
或是撥打免付費客服專線0800-818-777,由專人為您洽詢
-
家有遺傳疾病卻想懷孕怎麼辦☛超快速胚胎著床前基因檢測(UPGD)
-
擁有不孕困擾或是胚胎著床失敗的夫妻,可以參考這項檢測☛好孕到
遺傳疾病帶因篩檢常見問答
Q1:什麼是帶因者篩檢?
A:決定寶貝一出生的健康與否,往往來自於寶貝遺傳自父母親的基因。
目前已知人類有3萬多個基因,已知的遺傳疾病就已達一萬多種,其中又以隱性的遺傳疾病最為可怕,80%的患病孩童是沒有家族史,多半等到懷胎十月產下胎兒,並發現寶寶居然患有遺傳性疾病後,父母雙方才會意識到自身竟是帶有的致病基因的「帶因者」!
幸好,基因醫學運用於孕產前的第一步,可針對夫妻雙方身上各自的基因狀況做評估,針對下一代是否有重大殘疾或出生後夭折的嚴重的單基因隱性遺傳疾病,均可提早在懷孕前提早知道,以提早與專業醫師諮詢懷孕計畫。
Q2:一般帶因篩檢的疾病不是都很罕見,如果沒有家族史是否就沒有檢測帶因篩檢的必要性?
A:否。根據文獻資訊顯示,常規篩檢之胎兒異常發生率,如:唐氏症的發生風險平均為1/800、微小片段缺失與重複症候群和神經管缺損的風險均為1/1000。然,根據WHO統計,目前已知的單基因疾病的風險綜合發生率答1/100,因此單項疾病也許罕見,但綜合的發生率卻無法忽略! 且患病孩童,有80%均無任何的家族史,同步常規的產檢(血液、影像等)均難以發現,也因此僅能透過帶因篩檢才能及早發現此風險。
Q3.若經檢測我是帶因者,是否就代表下一代100%會患病?
A:否。當檢測出自身為體染色體隱性遺傳疾病的帶因者時,須另一半亦為此疾病帶因者時,才會造成下一代不分性別有高達25%患病的疑慮;然而,當為X染色體性聯遺傳疾病的帶因者,則只要媽媽是帶因者,下一代男性就有高達50%的患病風險,下一代女性則有50%與媽媽同為帶因者,且帶因者根據疾病的不同,也可能從沒有症狀到有輕微的臨床症狀。
也因此,通常在檢測帶因者時,會建議夫妻雙方同時檢測,才能儘早評估下一代的患病風險,或是若選擇僅夫妻其中一方受檢,則建議女方先受檢,才能評估到X染色體性聯遺傳類別的疾病。
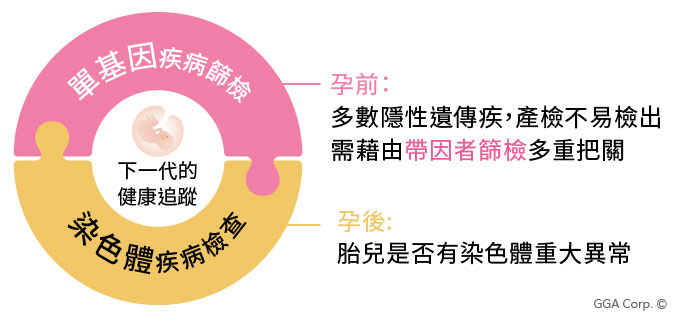
Q4:若我預備成功懷孕後,要檢測非侵入性染色體篩檢(NIPT/NIPS),或是抽羊水做羊水晶片以及核型分析,這些檢測工具是否可同步評估我是否為帶因者?
A:不行。非侵入性染色體篩檢(NIPT/NIPS)或是抽羊水做羊水晶片與核型分析,均屬於檢測染色體有無明顯的重大缺失,然而帶因者檢測的係屬於更小層次單一基因的隱性遺傳疾病,因此須透過其他的檢測工具做評估。
檢測染色體有無明顯的重大缺失,以及檢測有無單基因隱性遺傳疾病的風險,對於孕育下一代均至關重要,且檢測無法互相取代,就如同一個是用放大鏡看布料有沒有明顯的撕裂痕跡,另一項則是須要透過顯微鏡看布料纖維有無更細微的斷裂是放大鏡看不到的。
Q5:若檢測後夫妻雙方確實屬於高風險族群,下一步可以怎麼做?
A:檢測後若為高風險族群,建議先與醫師進行專業的遺傳諮詢。一般而言,透過夫妻雙方帶因的基因位點,可以在醫師的諮詢下,了解下一代若為患者,可能面臨的狀況與問題。
若夫妻雙方均尚未懷孕,可以選擇不以自然受孕的方式懷孕,改以人工生殖,利用胚胎著床前基因診斷技術(PGD),篩選沒有基因異常的胚胎植入母體,一方面降低流產機會,也同時避免懷上有遺傳疾病的胎兒;反之,若夫妻雙方已懷孕,則可以在專業醫師的指示下,選擇透過侵入性的方式,如羊膜穿刺、絨毛膜篩檢等方式,評估胎兒是否為患者。
Q6:如果孕前或懷孕期做了帶因檢查沒問題,是否可以取代寶寶出生後的新生兒篩檢呢?
A:否。任何的檢測項目從影像、血液一直到基因的檢測,均非百分之百保證寶貝一定健康。
舉例而言,產前已經有檢測過常見的遺傳性聽力損失的帶因檢測均父母雙方均無任何異常,然而小朋友的聽力問題,還是有可能有其他罕見基因上的疾病,會漸進型地影響聽力的狀況,甚至還有可能因其他後天性的因素而導致,包含感染、外傷、以及環境噪音等因素。也因此,產前的帶因檢測,無法取代寶寶出生後的常規新生兒篩檢!
遺傳疾病案例,聽聽醫師怎麼說





