量子力學計算軟體
既全面又精準的量子化學計算軟體
Materials Studio係提供基於密度泛函理論、QM/MM混合、以及半經驗等方法並給予使用者具高確效及友善介面的量子力學應用工具。量子力學方法提供正確的熱力、動力及結構優化結果,能有效幫助實驗進行全面的分析。此外,這些方法提供原子尺度的視角,讓使用者能了解物化反應機制是如何發生的。這些功能已經常應用在替代能源材料、催化物及半導體等材料開發上。
Materials Studio量子計算與催化工具可以正確預測以下性質,包含:
- 分子和晶體的幾何結構最佳化
- 化學反應路徑預測
- 光學性質
- 光譜(UV/Vis, IR, Raman, NMR, EELS, ELNES)
量子力學與化學、電性、光譜模擬包含以下模組:
Adsorption Locator
Adsorption Locator讓使用者可以模擬一個可負載固定成分的吸附物或混合物的基板。此獨特設計的系統主要能允許使用者找到吸附物在週期及非週期基板上最低能量的吸附位置,或像是研究複合吸附物的吸附行為。利用模擬溫度慢慢降低下之基板-吸附物系統的組態空間來作Monte Carlo搜尋,Adsorption Locator可以找出所有在該條件下可能的吸附組態,對過渡態計算與反應性吸附的初始結構建立非常有幫助。
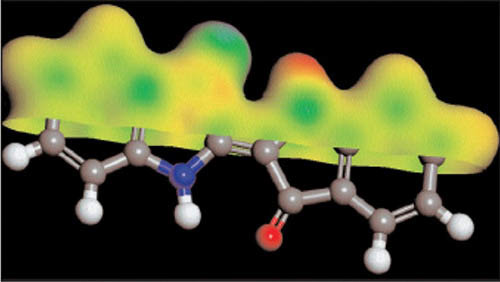
CASTEP
在模擬晶格結構的軟體中,CASTEP是首屈一指被廣泛運用的量子計算程式。CASTEP最初是由劍橋大學的凝聚態物體團隊所發展,它使用密度泛函理論去模擬各種固態材料,晶格介面表面的性質。CASTEP是基於全能量平面波膺勢方法並適用於週期系統,可預測包含晶格常數、分子幾何結構、結構性質、能量帶結構、態密度、電荷密度和光學等性質。具效率的平行版本程式能模擬幾百顆原子的大系統。
DFTB+
DFTB+(Density Functional based Tight Binding)是一以於緊束縛量子力學為基礎的獨特密度泛涵理論,它能讓使用者研究團簇、分子、固態和奈米結構。DFTB+合併精準且具可性度的DFT計算和簡潔具有效力的緊束縛法來提供對穩定態與激發態性質的計算。被用來將模型的電性與排斥能量貢獻參數化,可產生為擬合短程排斥勢能的Slater-Koster檔案,以近似的方法來來處理大系統的分子電性計算。
DMol3
DMol3是一款以密度泛函理論做為基礎的量子力學程式,它支援週期與非週期系統,提供使用者研究關於氣相、溶劑、表面和固態環境等模擬領域。由於DMol3對靜電力的獨特計算方法,讓DMol3一直以來成為結構分子系統最佳化中最快速的密度泛函計算方法之一。DMol3也可以結合LST/QST算法和共軛梯度限制做有效率的過渡態計算,因此可以避免展開繁複的Hessian矩陣,進一步增加計算效率。

Gaussian MS User Interface
Materials Studio Gaussian 2003使用者介面讓你可以透過Gaussian 03 server執行Hartree-Fock (HF)和density functional theory (DFT)的運算。利用Gaussian,你可以預測結構、電子性質、熱力性質、NMR與震動光譜。MS Gaussian 2003使用者介面可以藉由Job File option執行Gaussian 03所有的功能,需注意的是使用者必須要另外購買Gaussian 03軟體以便完善所有功能。
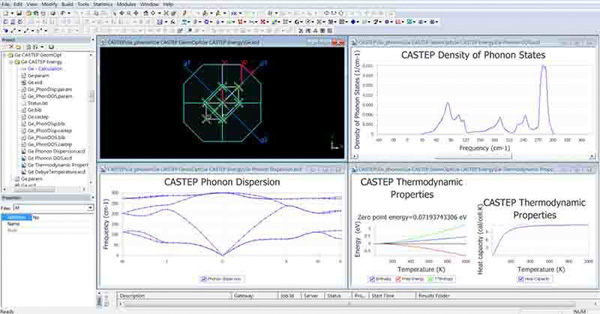
NMR CASTEP
NMR CASTEP為CASTEP模組的進階版本。它包含全部CASTEP的功能之外,並另外增加正確NMR (Nuclear Magnetic Resonance Spectroscopy) 化學位移張量、等向位移與任何微量材料相對等向位移的電場梯度之預測功能。計算化學位移讓晶格多組態之實驗結果可用此模組明確的區別及分析,找出無機晶體結構的亂序程度。NMR CASTEP亦可被應用去計算各種材料形態,如分子、固體、材料間界面和表面。而材料種類包含有機分子、陶瓷和半導體皆能使用此模組來有效分析。
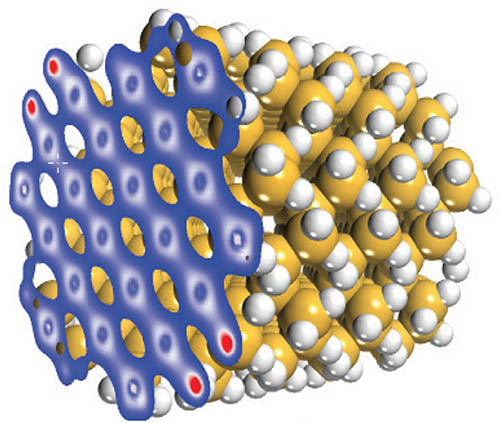
-ONETEP
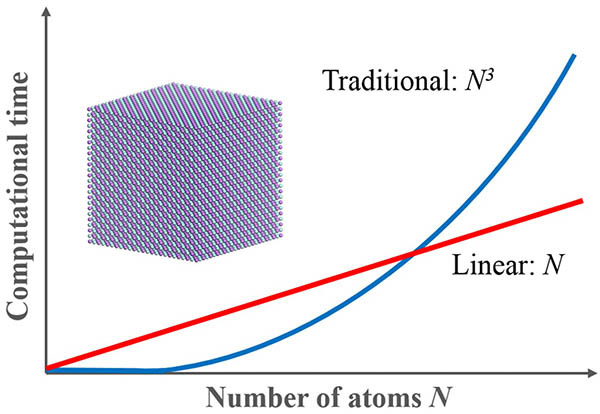
-ONETEP是一個具革新意義的量子力學程式,它主要是設計用於計算大系統,可有效的增加計算效率。
-ONETEP基於密度泛涵理論為主的線性伸縮方法,通常需要的計算時間會隨著原子數量呈三次方倍的增加,但-ONETEP能讓計算時間以線性增加,大幅減少計算資源的消耗。因此它非常適合在大分子系統的表面化學、結構性質和組態的研究上,如應用在計算半導體和陶瓷材料的缺陷性質(空缺、隙縫、置換型雜質、晶界與錯位)。
QMERA
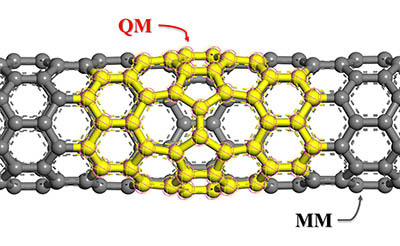
QMERA能讓使用者結合量子力學(quantum mechanical, QM)和分子力學(molecular mechanics, MM)計算在非週期系統使用ChemShell環境。ChemShell為一計算化學環境,基於Tcl 語言處理混合QM/MM的程序。QMERA的特點是可以將部分所選擇需要著重及電子性質的計算使用QM,而其他則使用MM配合對應的勢能做分子力學的計算,如此一來便能減少計算時間,可獨立呈現關鍵部分的計算結果。
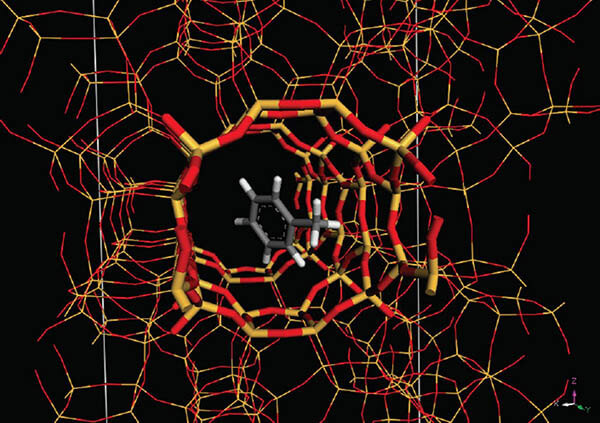
Sorption
Sorption模組讓使用者可以模擬純的被吸收物(或是混合成份的被吸收物)在吸收骨架中被吸收,也就是建構一個具適合尺寸及形狀之孔洞的的三維周期結構對被吸收物分子。典型吸收劑的範例包含微觀孔洞與界觀孔洞材料,像是沸石、鋁矽酸鹽、泥、奈米管、高分子薄膜、矽膠、活性碳和有機金屬骨架。Sorption可以提供吸附等溫、鍵結點位、鍵結能、密度和能量場、能量分佈、吸收選擇性、溶解度參數、等量吸收熱和亨利常數。除此之外,Sorption模組亦可輸出各種性質且符合實驗結果的資料,可直接比對實驗與計算結果上的差異。

VAMP
VAMP是最先進的半經驗量子力學程式,可模擬氣相與溶液相分子的反應和性質。程序具有高精度的穩定與快速計算的優勢,甚至在大分子系統也可以有非常好的效率去呈現。
VAMP特別加強幾何結構最佳化和過渡態與電子性質的計算部分,包含模擬溶液效應和計算多性質如極矩、極化率、電荷密度、靜電位能、熱力性質以及碳13化學位移等性質。它提供的各種半經驗勢能,包含AM1、PM3與PM6等勢能皆有相當高的準確性。