Polymers and Classical Simulation Software
The classical mechanics simulation software
Materials Studio's polymer and simulation modeling software allows you to construct and characterize models of isolated chains or crystalline and amorphous bulk polymers. Its prognostic abilities enable you to predict key properties, including miscibility and blending, cohesion and wetting, mechanical behavior, diffusion, and adhesion at surfaces. Applications include paints and coatings, lubricants, food packaging, gels, and adhesives.
The Modules of Polymers and Classical Simulation Software are shown as follows:
Amorphous Cell
Amorphous Cell is a suite of computational tools that allow you to construct representative models of complex amorphous systems and to predict key properties. Among the properties that you can predict and investigate are cohesive energy density, equation-of-state behavior, chain packing, and localized chain motions. The methodology of Amorphous Cell construction is based on an extension of well-established methods for generating bulk disordered systems containing chain molecules in realistic equilibrium conformations. Other features include provision for construction of arbitrary mixture systems containing any combination of small molecules and polymers, in addition to special capabilities for producing ordered nematic mesophases and slabs of amorphous material suitable for use in creating models of interphases, as would be required to study adhesion or lubrication.

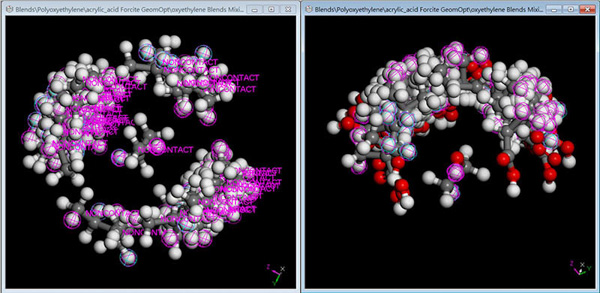
Blends
Blends combines a modified Flory-Huggins model and molecular simulation techniques to calculate the compatibility of binary mixtures. These mixtures range from small models to large systems, including polymer solutions, polymer blends, and alloys. The information obtained includes phase diagrams (critical points, binodal, and spinodal curves), thermodynamic mixing variables (energy and free energy of mixing), the temperature-dependent interaction parameter χ, binding energy analysis, and the identification of favorable binding configurations between molecular pairs, molecules and surfaces, additives and bulk materials, and liquids and crystals.
COMPASS
COMPASS is a powerful forcefield supporting atomistic simulations of condensed phase materials. COMPASS stands for condensed-phase optimized molecular potentials for atomistic simulation studies.
COMPASS is the first ab initio forcefield that has been parameterized and validated using condensed-phase properties, in addition to various ab initio and empirical data for molecules in isolation. Consequently, this forcefield enables accurate and simultaneous prediction of structural, conformational, vibrational, and thermophysical properties for a broad range of molecules in isolation and in condensed phases, and under a wide range of conditions of temperature and pressure.
Conformers
The Conformers module provides methods for searching the conformational space of nonperiodic molecular systems in order to derive a reasonable sampling of the low energy conformations. The primary degree of freedom explored is the set of rotatable torsion angles of the molecular system. The Conformers module can perform both systematic and stochastic (random) searches.
Once you have performed a Conformers run and obtained some conformers, the Conformers module allows you to calculate a range of descriptors, enabling you to perform a detailed analysis of the structures. Conformers also gives you the option of performing an extended search on the output from a run. You can select a subset of the conformers produced by an initial Conformers run and carry out a secondary search around each of these structures.
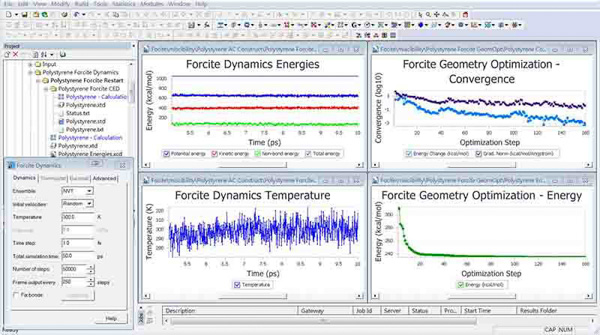
Forcite
Forcite is a molecular mechanics module for potential energy and geometry optimization calculations of arbitrary molecular and periodic systems using classical mechanics. Forcite offers support for the COMPASS, UFF, and Dreiding forcefields. With this wide range of forcefields, Forcite can handle essentially any material. The geometry optimization algorithm offers steepest descent, conjugate gradient, and quasi-Newton methods, in addition to the Smart algorithm, which uses these methods sequentially. This allows very accurate energy minimizations to be performed.
Forcite Plus
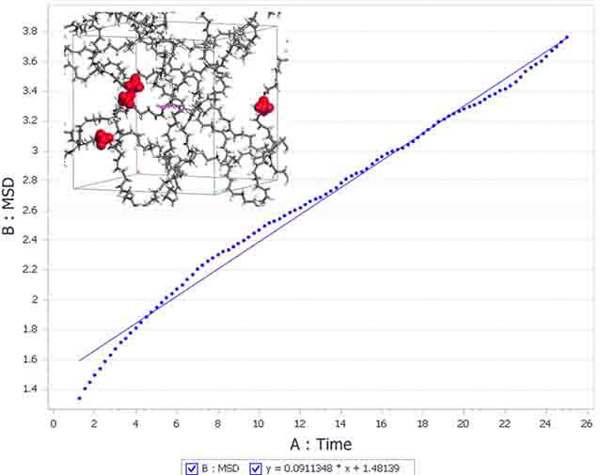
Forcite Plus is an enhanced version of the Forcite module. It includes all of the Forcite functionality and extends it to include molecular dynamics and analysis. The addition of molecular dynamics to Forcite Plus allows the study of time-dependent properties such as diffusivity. A comprehensive suite of analysis tools allows the modeler to analyze simple properties, such as density variations, and calculate complex properties, like dipole autocorrelation functionals.
Forcite Plus can work with isolated molecules, 2D surfaces, and 3D periodic systems. This allows you to perform molecular dynamics on systems ranging from an organometallic complex to a catalyst surface, or even a bulk amorphous polymer.
GULP
The General Utility Lattice Program (GULP) is designed to perform a variety of tasks relating to three-dimensional solids. Although it started life as an attempt to produce an input file-driven program for interatomic potential fitting, it has now expanded to encompass energy minimization, phonon calculations, and other useful facilities. GULP can also perform calculations on nonperiodic systems. This facility is useful when calculating defect energies for molecular defects. It also allows the combined fitting of potentials to bulk and cluster information.
Mesocite
Mesocite is a molecular mechanics module for potential energy, geometry optimization, and classical dynamics calculations. Periodic and nonperiodic mesoscopic systems can be studied as beads representations, where each bead corresponds to a collection of atoms. Mesocite offers support for the MS Martini and Shinoda2007 forcefields. The geometry optimization algorithm offers steepest descent, conjugate gradient, and quasi-Newton methods, in addition to the Smart algorithm, which uses these methods sequentially. Classical dynamic trajectories can be calculated for a range of conditions by the application of a number of thermostats and barostats.
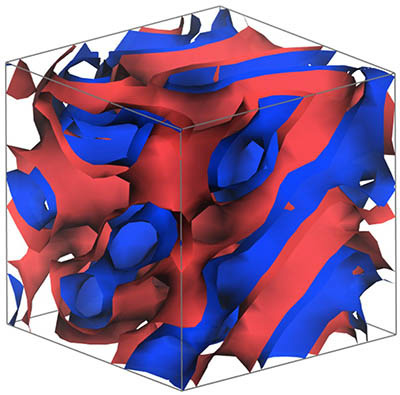
MesoDyn
MesoDyn is a dynamic mesoscale method for the study of large systems over long time scales. The algorithm dynamically evolves component density fields, driven by both chemical potential gradients and by Langevin noise. The species in the system interact through effective external potentials derived from the calculated energies of mixing or Flory-Huggins parameters. Atomistic detail is sacrificed in favor of larger systems and longer times.
Phenomena such as microphase separation, micellar aggregation and self-assembly can be readily studied with MesoDyn. The effects of shear and confinement in fixed geometries can be investigated.
The applications of MesoDyn include: paints and emulsions, cosmetics and personal care, blended polymeric materials, surfactant solutions, complex drug delivery systems, and many others.
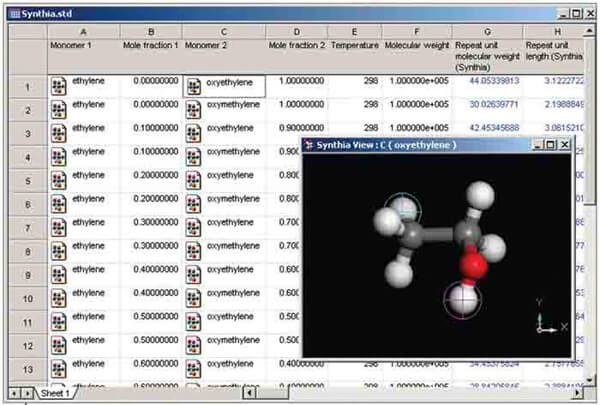
Synthia
The Synthia module allows you to make rapid estimates of polymer properties using empirical and semiempirical methods. Synthia can predict a wide range of thermodynamic, mechanical, and transport properties for bulk amorphous homopolymers and random copolymers. The key advantage of Synthia is that it uses connectivity indices, as opposed to group contributions, in its correlations; this means that no database of group contributions is required and properties may be predicted for any polymer composed of any combination of the following nine elements: carbon, hydrogen, nitrogen, oxygen, silicon, sulfur, fluorine, chlorine, and bromine.
This methodology is based on research conducted by Dr Jozef Bicerano (2002).