Quantum and Catalysis Software
The Comprehensive Quantum software!
Materials Studio offers validated, efficient, and user-friendly quantum mechanical applications based on Density Functional Theory (DFT), hybrid QM/MM and semi-empirical methods. Quantum mechanical methods yield accurate thermodynamic, kinetic, and structural results, providing an efficient adjunct to experiment. In addition, the methods provide insight into processes at the atomic level, allowing you to understand why and how a process occurs. Applications include alternative energy materials, catalysis, sensors and semiconductors.
Materials Studio quantum and catalysis tools accurately predict:
- Molecular and crystal geometry
- Chemical reaction pathways
- Optical properties
- Spectra (UV/Vis, IR, Raman, NMR, EELS, ELNES)
The Modules of Quantum and Catalysis Software are shown as follows:
Adsorption Locator
Adsorption Locator enables you to simulate a substrate loaded with an adsorbate or an adsorbate mixture of a fixed composition. Adsorption Locator is designed for the study of individual systems, allowing you to find low energy adsorption sites on both periodic and nonperiodic substrates or to investigate the preferential adsorption of mixtures of adsorbate components, for example.
Adsorbates are typically molecular gases or liquids and substrates are usually porous crystals or surfaces, such as zeolites or carbon nanotubes, or amorphous structures, such as silica gel or activated carbon. Adsorption Locator identifies possible adsorption configurations by carrying out Monte Carlo searches of the configurational space of the substrate-adsorbate system as the temperature is slowly decreased.
CASTEP
Originally developed in the Theory of Condensed Matter Group at Cambridge University, UK, CASTEP is a program that employs density functional theory (DFT) to simulate the properties of solids, interfaces, and surfaces for a wide range of materials classes. Based on total energy plane-wave pseudopotential methods, CASTEP takes the number and type of atoms in a system and predicts properties including lattice constants, molecular geometry, structural properties, band structures, density of states, charge densities and wavefunctions, and optical properties. Efficient parallel versions of the code are also available to simulate large systems containing hundreds of atoms.
DFTB+
DFTB+ is a unique density functional based tight binding quantum mechanical code that allows users to study clusters, molecules, solids, and nanostructures. DFTB+ merges the precision and reliability of DFT with the simplicity and efficiency of tight binding, offering universality for ground-state and excited-state property calculations. Slater-Koster files are used to parameterize the electronic and repulsive energy contributions to the model.
DMol3
DMol3 is a unique density functional theory (DFT) quantum mechanical code that allows users to study problems in the gas phase, solvent, surface, and solid environments. Owing to its unique approach to electrostatics, DMol3 has long been one of the fastest methods for molecular DFT calculations and can quickly perform structure optimizations of molecular systems using delocalized internal coordinates. DMol3 can also be used to search very efficiently for transition states using a combination of LST/QST algorithms with conjugate gradient refinement, thereby avoiding the computationally expensive calculation of the Hessian matrix.

Gaussian MS User Interface
The Materials Studio Gaussian® 2003 User Interface allows you to access the Gaussian 03 server for the study of molecules using Hartree-Fock (HF) and density functional theory (DFT) methods. With Gaussian, you can predict structure, electronic properties, thermodynamic properties, NMR and vibrational spectra.
The MS Gaussian® 2003 User Interface currently only exposes a limited subset of Gaussian 03 functionality. However, by using the Gaussian Job Files option, the full functionality of Gaussian 03 can be accessed.
Gaussian is a registered trademark of Gaussian, Inc.
The Gaussian 2003 server application is not included in the Materials Studio installation and must be purchased separately from Gaussian.
-ONETEP
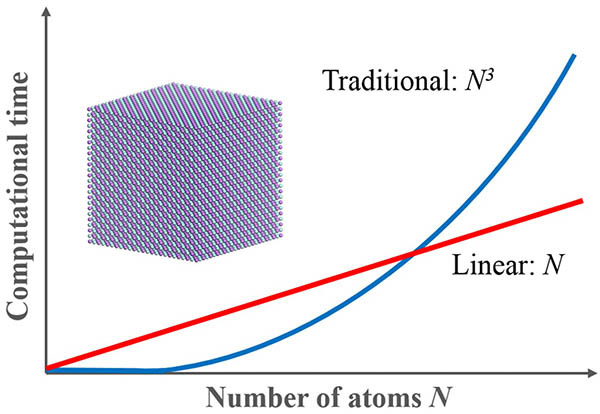
-ONETEP is a revolutionary quantum mechanics-based program designed specifically for calculations on large systems. -ONETEP employs density functional theory (DFT) in the density matrix formulation and is a linear scaling method, that is, the time required for the total energy calculation increases linearly with the number of atoms. As a result, -ONETEP is particularly well-suited to application in the study of surface chemistry, structural properties, and the configurations of large molecular systems. -ONETEP can also be used to study the properties of defects (vacancies, interstitials, substitutional impurities, grain boundaries, and dislocations) in semiconductors and ceramic materials.
QMERA
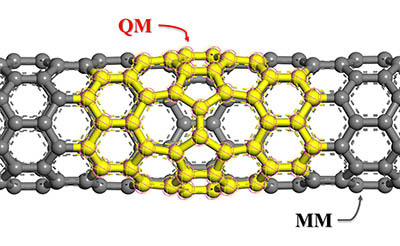
QMERA is a program that allows you to perform combined quantum mechanical (QM) and molecular mechanics (MM) forcefield calculations on nonperiodic systems using the ChemShell environment. ChemShell is a computational chemistry environment, based on the Tcl interpreter, that takes over the communication and data handling for hybrid QM/MM calculations, leaving the time-consuming energy evaluations to specialized external codes (visit the ChemShell home page for further details).
Sorption
The Sorption module allows you to simulate a pure sorbate (or mixture of sorbate components) absorbed in a sorbent framework, that is, a three-dimensional periodic structure with pores of a size and shape suitable to accommodate the sorbate molecules. Typical examples of sorbents include microporous and mesoporous materials such as zeolites, aluminophosphates, clays, nanotubes, polymer membranes, silica gels, activated carbons, and metal-organic frameworks. Sorption simulations can provide adsorption isotherms, binding sites, binding energies, density and energy fields, energy distributions, sorption selectivities, solubilities, isosteric heats, and Henry constants.
The Sorption module has been designed to be used by experimentalists as well as computational chemists. Output-analysis features such as automatic calculation and display of isotherms make it easy to directly compare Sorption data with experimental results.

VAMP
VAMP is a state-of-the-art semiempirical quantum mechanics program that simulates reactions and properties of molecules in gas and solvent phases. The program is optimized to be highly numerically stable and fast so that calculations, even on large molecular systems, can be performed very efficiently.
VAMP contains many enhancements to geometry and transition state optimization and electrostatics. It simulates solvent effects and calculates many properties such as dipole moments, polarizabilities, charge densities, electrostatic potentials, thermodynamic properties, and 13C chemical shifts.