

BIOVIA Discovery Studio
生物模擬軟體簡介
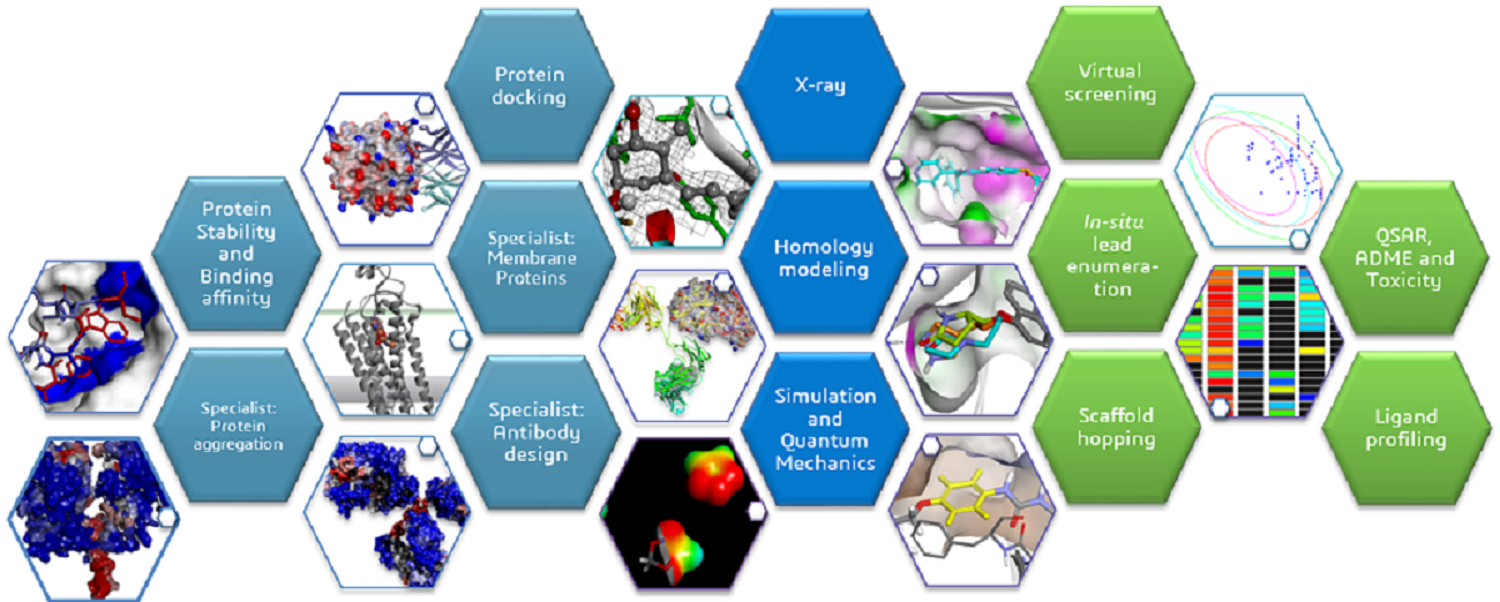
應用領域
BIOVIA Discovery Studio是Dassault Systèmes公司針對生命科學領域所開發,一個功能強大的研究平臺,整合了廣泛的生物與化學資訊學、分子模型建造和模擬工具。常應用於蛋白質體、AI藥物開發過程與研究,為科學家提供易用的蛋白質序列比對、優化、AI藥物開發設計工具。通過高品質的圖形、多年驗證的技術集成的使用環境,Discovery Studio可將實驗資料的保存、管理與專業水準的建模、模擬工具整合為一,提升研究團隊合作開發的溝通效率與品質。
使用介面及擴展性
BIOVIA DS是建立在最新的流程管理平臺BIOVIA Pipeline Pilot之上,其高度彈性的流程管理介面讓資料的共用和交流變得更為方便和簡潔,同時這個開放平臺技術也保留了讓使用者整合自己的或第三方的軟體工具的可能性。研究人員可以在一個統一的平臺上完成從基因到先導化合物設計之一系列工作。也可以直接使用Perl腳本語言來執行相關功能。此外,DS為一個用戶端-伺服器(Client-Server)架構之平臺,其用戶端的視覺化程式可讓研究人員在普通個人電腦上操作使用以上工具。
主要模擬功能
生物大分子 (Biologics)
提供以下模擬應用工具:
- 蛋白/核酸的序列分析
- 大分子三維結構建模
- 蛋白質結構性值計算
- 蛋白突變設計
- 虛擬胺基酸突變
- 雙硫鍵預測
- 蛋白聚集效應預測
- 抗體設計與優化
- 抗體人類化
- 抗體親和力計算
- 蛋白質-蛋白質相互作用預測
- X-Ray晶體結構解析
- 分子動力學模擬和QM/MM等。
化合物小分子 (Small Molecules)
提供以下模擬應用工具:
- 化合物虛擬篩選
- 全新化合物設計
- 片段生長、骨架取代
- 受體-配體作用機制解釋
- 分子對接
- 基於片段的藥物設計及優化
- 結合能自由能計算
- FEP自由能微擾
- 基於結構/化合物之藥效基團模型建立
- 化合物數據庫建立和篩選
- 藥效基團數據庫建立
- QSAR/QSPR、活性關係分析(MMP)
- 化合物構型/性質篩選和分析
- 化合物ADMET性質預測等
基本和視覺化界面
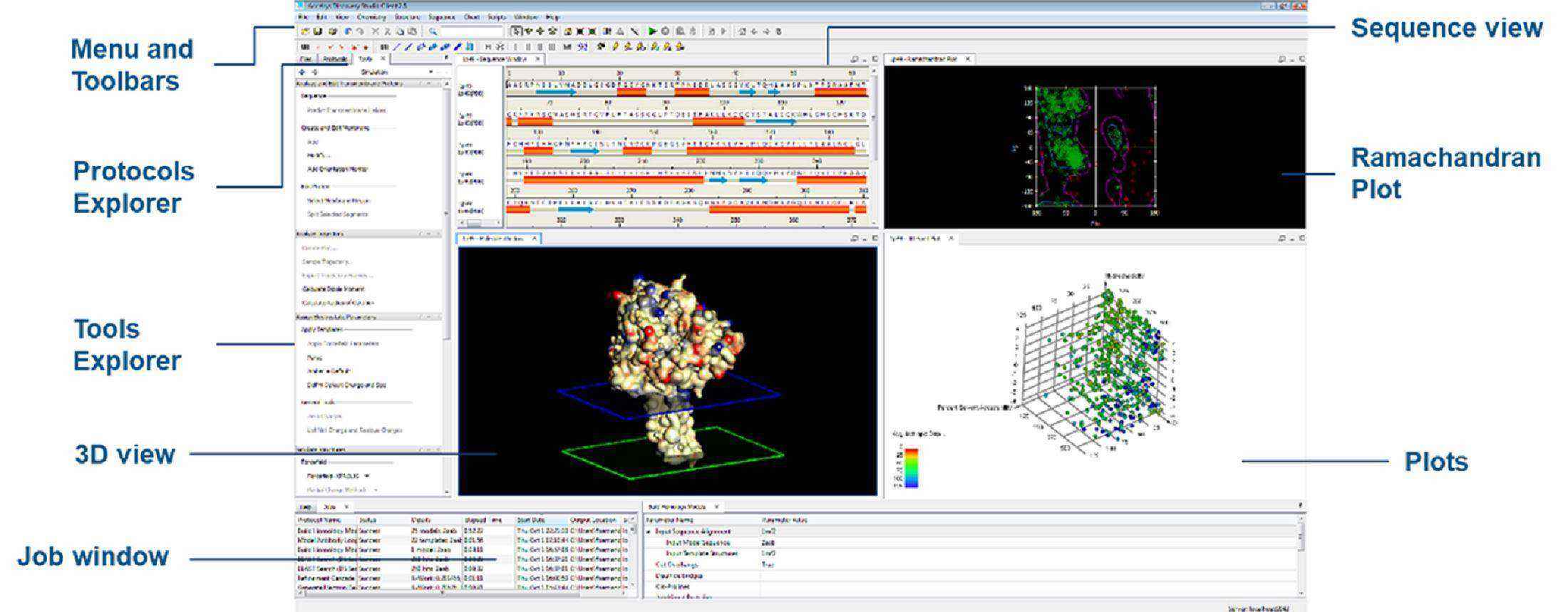
DS Client
客戶端的視覺化界面,作為圖形化視窗的服務平台,必須與伺服器和權限連線設定後,才能啟用完整之功能。可與BIOVIA Pipeline Pilot伺服器共同使用,為使用者提供數據、工作流程和計算資源共享。
BIOVIA Pipeline Pilot Server
作為客戶端(Client)-權限(License)-伺服器(Server)模式中的軟體主要計算伺服器端。
BIOVIA Draw
免費的分子二維結構顯示套件。可用於Windows環境,使用者能夠輕鬆地繪製和編輯複雜的分子,化學反應和生物學序列,從而促進了對科學信息的協作搜索、查看、交流和存檔。
蛋白質建模和模擬

可利用未知結構之蛋白質序列與其他已知具同源性之蛋白質結構,自動預測蛋白質結構,為一快速且標準化之建模方法。可預測類型包含激酶、抗體、穿膜蛋白等。建模模組結合蛋白質/核酸的序列分析功能,可自動化完成抗體Fab、Fv、全長及雙特異性結構之模擬。
Search Sequence by Similarity/Align Sequence and Structure
可利用BLAST及PSI-BLAST演算法來搜尋本地端資料庫或NCBI資料庫,確認蛋白質序列之同源區域。DS同時也支援抗體序列之識別功能,計算蛋白質分子量、等電點、淨電荷等生物物理學性質。
Analyze Sequences/Protein Conservation Pattern
分析蛋白質家族之序列、三級立體結構之保守區域及胺基酸之位置,可快速了解蛋白質功能機制。
Prepare Protein/Verify Model
蛋白質立體結構之評估工具,可運用資料庫或Profiles 3D方法協助使用者快速找出蛋白質結構中不合理之區域。
Create Homology Models
結合MODELER和CHARMm模組,可計算抗體人類化,自動預測並提供抗體序列中可人類化突變的殘基位置及後續突變資訊,協助減少抗體的變異性並同時確保結構穩定性及抗原結合的特異性。
Minimize and Refine Protein
利用CHARMm對建模之蛋白質結構進行側鏈及loop區之優化,提高同源蛋白質模型之準確性。
Dock and Analyze Protein Complexes
快速且準確的蛋白質-蛋白質(抗體/抗原)之結構對接預測工具。DS結合廣泛被使用之ZDOCK、ZRANK及RDOCK三個模組,ZDOCK用於蛋白質-蛋白質對接,ZRANK用於分級ZDOCK之能量,RDOCK則基於CHARMm能量最小方化之方法優化結構。
Predict Protein Formulation Properties
可用於建購及修飾蛋白質、胜肽分子、蛋白質複合物之組成,快速產生蛋白質性質報告,計算蛋白質溶劑能、pKa值、pH變化及溶解性和黏性。
預測抗體、蛋白質、多胜肽等生物類型的藥物在製造、生產過程的聚集效應,進而確定其穩定性。可通過預測影響蛋白質之胺基酸位點,來進行胺基酸突變以協助蛋白質設計及優化。
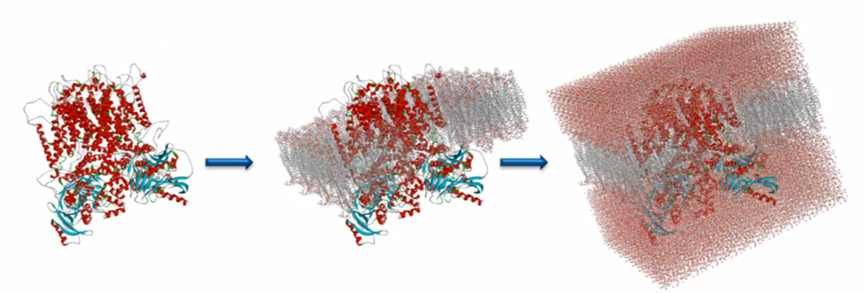
分子力學、分子動力學、分子力學/量子力學之模擬
DS Molecular Dynamics Tools
結合工業標準之分子力學及動力學之步驟。利用經典且強大的CHARMm計算模組及力場,進行分子動力學模擬,動態的研究蛋白質、核酸、醣類、脂質、小分子等複合物於不同環境和狀態下的熱力學/動力學特性。同時DS整合NAMD分子動力學模組,加快計算速度。目前DS2021最新版更支援GPU加速運算功能,NAMD計算為8個CPU平行運算之9倍速度。另外,為了研究時間尺度較長之生物學過程,DS CHARMm支援分子拉伸動力學模擬,可動態研究配體的結合過程、通道蛋白中配體之分離過程及蛋白質去摺疊化過程及結構變化,以預測受體/配體結合自由能。
Analyze Trajectory
分子動力學結果分析和視覺界面。能夠分析和呈現分子動力學分析軌跡文件,包含主成分分析、計算路徑分布函數、原子間距離和角度等變化分析、RMSD和RMSF分析等。
DS Quantum Mechanism
使用量子力學/分子力學(QM/MM)方法,從電子面向優化標靶蛋白-配體複合物之結合位的構型、能量及力學計算。其中量化方法採用基於密度泛函理論的DMOL3,力學優化採用CHARMm。
Run Simulation with Different Force Fields
Chemistry at HARvard Macromolecular Mechanics,為一分子模擬程序,廣泛被應用且具有完善的能量功能集,多種增強的採樣方法之多粒子系統,並支持包括QM / MM,MM / CG和多種隱式溶劑模型在內的多尺度技術。
主要針對生物系統,包括肽,蛋白質,輔基,小分子配體,核酸,脂質和碳水化合物,因為它們存在於溶液,晶體和膜環境中。 CHARMm還發現無機材料在材料設計中的廣泛應用。 可在包括平行集群和GPU在內的各種平台上實現高性能。
支持多種力場,包含CHARMm、CHARMm Polar H、charmm36、charmm27、charmm22、charmm19力場,可模擬多種Poisson-Boltsman(PB)、Generalized Born(GB)類型的溶劑模型。
DS CFF
第二類原子力場,可以應用於蛋白質、核酸、醣類、脂質、胜肽等大部分大分子及小分子結構之模擬研究。
DS MMFF
Merck Molecular Force Field,計算和實驗數據的第二類力場,可準確處理結構和非鍵結之交互作用,用於配體或有機小分子的研究。
基於結構之藥物設計
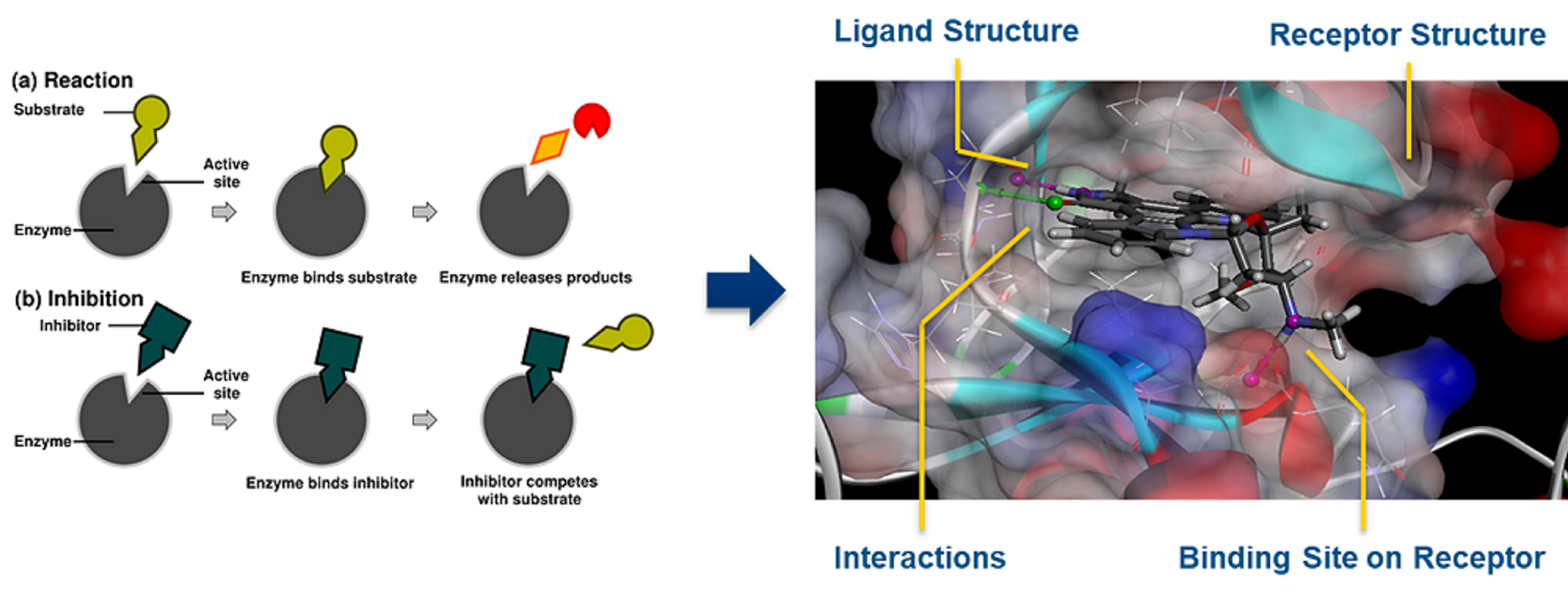
DS Flexible Docking
受體彈性對接工具(“Induced-Fit”對接工具)。可運用於配體及受體之雙彈性對接,模擬”誘導-契合”效應。其最大優勢為準確,可研究配體-受體交互作用之理論,適合於作用機制研究。
DS Libdock
快速的分子對接工具,適合於大規模資料庫進行之虛擬藥物篩選。速度快,並支援平行化運算。
DS CDOCKER
基於CHARMm的對接程序,採用soft-core potentials以及optional grid representation將配體與受體活性位點進行對接。將小分子各狀態之構型在受體活性位點區域進行優化,使對接計算結果更加準確。CDOCKER在對接計算中還可考慮藥效基團之限制,以提高效率和準確度。
DS GOLD interface
DS提供GOLD模組的串接,並提供設置interaction filter之功能。但使用者須有GOLD軟體之授權。
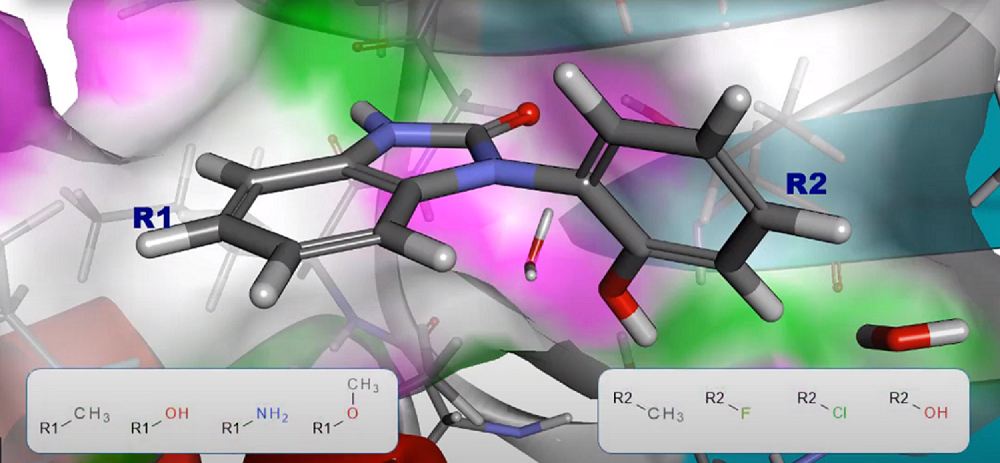
基於片段之藥物設計
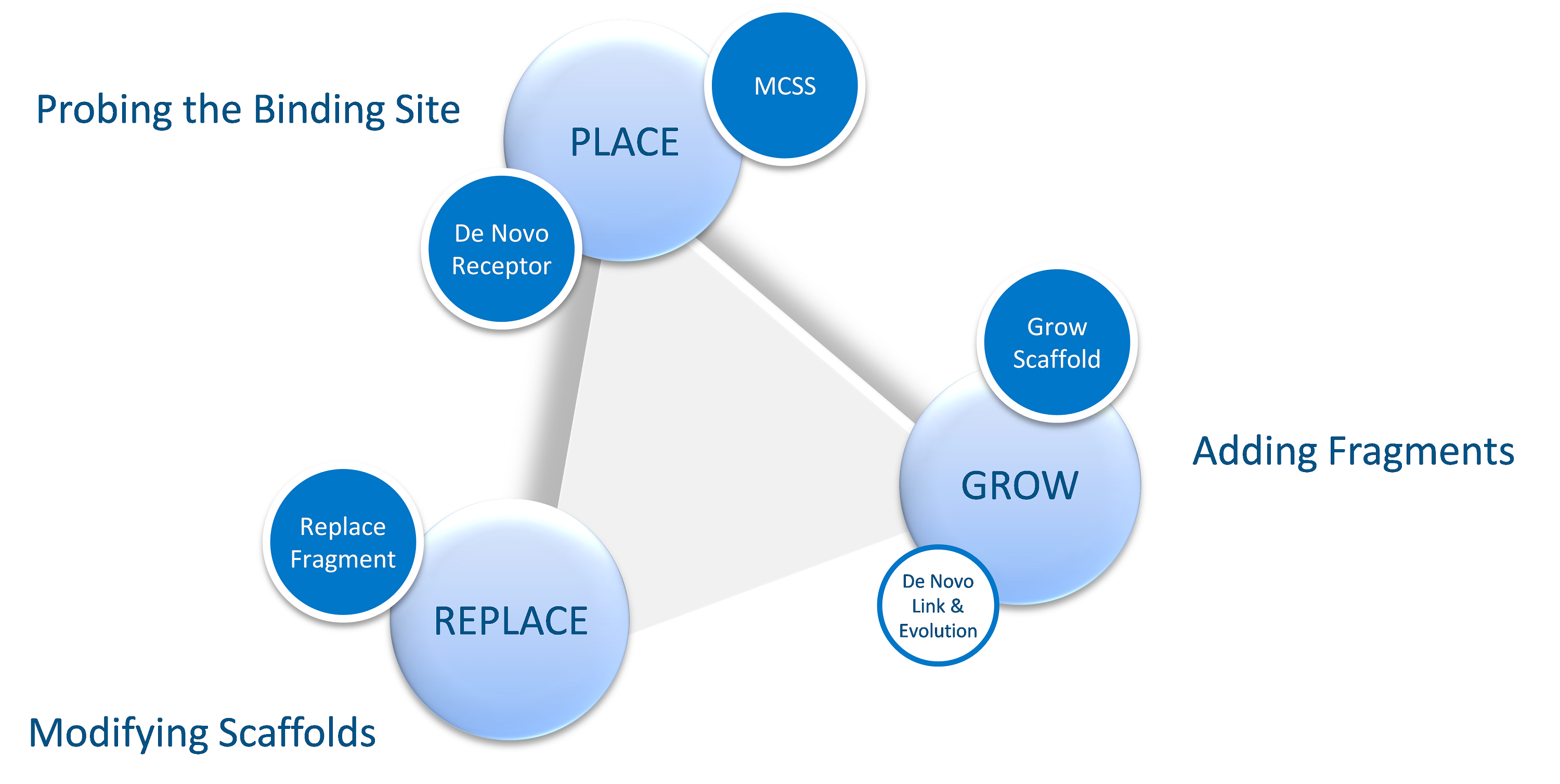
DS De Novo Evolution
以Ludi算法為基礎發展出的藥物設計方法。給予核心結構後,可自動化設計潛在之全新小分子化合物,大幅縮短藥物開發及改造的週期,為設計Me-Better類藥物分子之工具。
DS MCSS
基於片段的小分子藥物設計工具。可將多個分子片段同時對接進入蛋白質結合位置,尋找片段的最佳結合區域,也可以用來表現及分析蛋白質結合位點的特性。
DS Grow Scaffold
基於化學反應之生長分子片段之工具。能夠選取配體分子中特定原子或是特定基團作為反應位點,在選取經典化學反應進行片段生長,如: 醯胺合成反應、醚合成反應等。可選擇性採用MM-GBMV/SW模型優化配體分子或蛋白質側鏈。最後根據蛋白質配體特性進行Pareto優化,使得到之分子更易於實驗合成。
DS Replace Fragment
基於電子替換的骨架設計工具。可於受體-蛋白質結構或是化合物結構下進行,根據蛋白質口袋特性進行化合物修飾及Pareto優化。
基於藥效基團之藥物設計
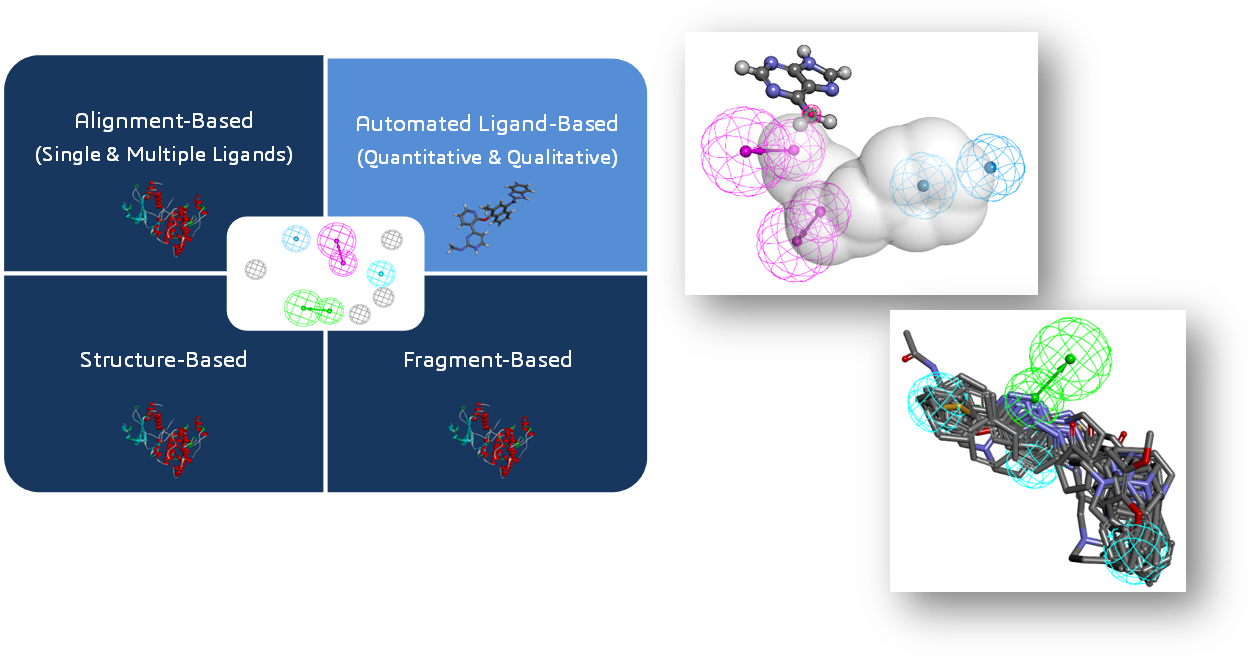
3D QSAR Pharmacophore
以一組化合物基於特性/相似性比對並自動生成藥效基團模型的工具。這些特型結構包含親/疏水性、氫鍵受體/給體、正/負電荷、芳香環及非鍵結之基團等。可選擇性生成/排除特性,以提高模型之預測能力,也可根據已知化合物知活性數據,建立3D-QSAR模型以預測新化合物活性。
Interaction Pharmacophore Generation
基於受體與配體結構產生之精準藥效基團模型。可以使分子滿足化學特性及受體結合位之互補特性,並探討受體間之障礙空間,以精準預測新化合物之活性作用位置。
Build Database
建立及管理In-House立體藥效結構資料庫,用於藥效集團模型的化合物虛擬篩選,協助使用者快速找出可能的先導化合物,並結合特性、形狀及特徵作為查詢條件。
DS De Novo Ligand Builder
藥效基團為主的全新分子建構工具。DS採用新穎而獨特的基於片段之藥物設計方法(FBDD),片段位置受藥效基團之影響,產生的化合物不僅能與蛋白質活性位點互補,化合物之間的互補性更是有利於產生新型先導化合物。
DS PharmaDB
藥效基團資料庫,BIOVIA Discovery Studio2021包含scPDB(2017)中16,034個複合物晶體結構、4,782個蛋白質及6,326配體所建構出25萬多個藥效基團模型,並根據不同的蛋白質類型進行了分類,是目前市場上最大的受體-配體複合物藥效基團資料庫。結合PharmaDB及DS之藥效基團功能模組,可以快速及有效進行標靶蛋白預測、中草藥複方成分作用蛋白預測,以及化合物副作用評估。
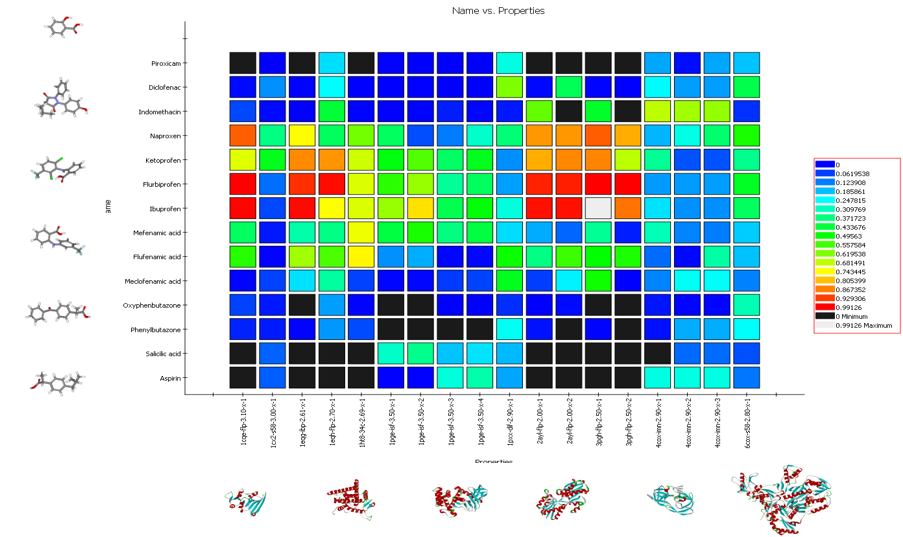
基於小分子之藥物設計
Create QSAR Model
量構效關係是一種藉助分子的理化性質參數或結構參數,以數學和統計學手段定量研究有機小分子與生物大分子相互作用、有機小分子在生物體內吸收、分布、代謝、排泄等生理相關性質的方法。DS可以計算千種與生物活性或ADME/T性質相關之化合物,包含分子拓譜描述、分子指紋在內的基本性質,並提供多種統計工具,如Bayesian模型、多元線性回歸、最小二乘法等,可協助於各種複雜資料分析及挖掘。
Study SAR
可利用已知活性、實驗數據快速獲取活性懸崖(cliff),近一步分析已知化合物之間的活性差異來設計新穎化合物。
Calculate Molecular Property using DMOL3 Descriptor
使用基於泛含密度理論的量子力學DMOL3計算分子與電子相關的描述。
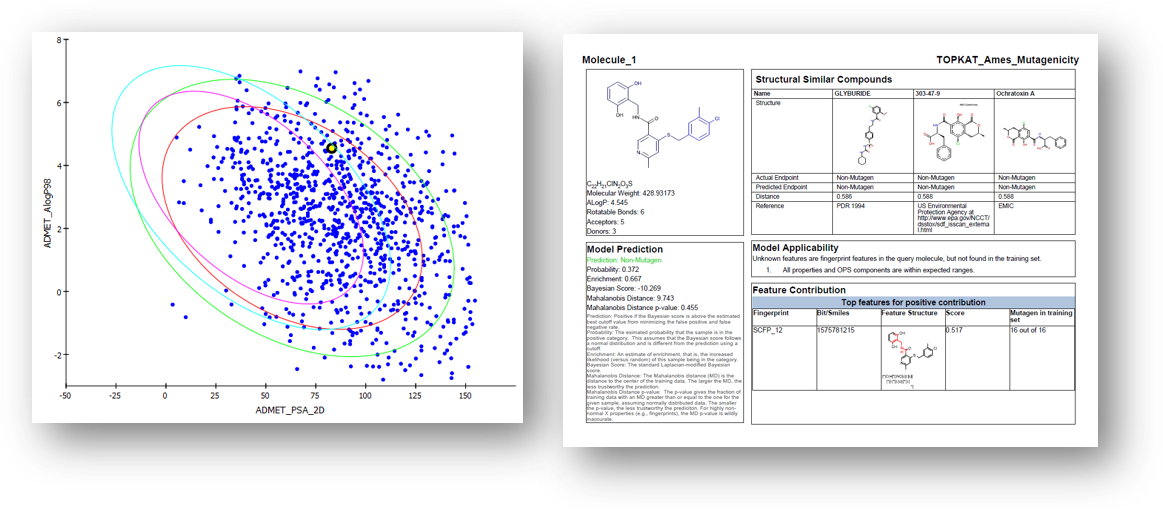
ADMET & TOPKAT Toxicity Prediction
提供從化學結構預測吸收、分布、代謝、排泄(ADME)和毒理性質的工具。ADMET現在包含了人體腸道吸收、水溶性、血腦屏障滲透、血漿蛋白結合、CYP2D6結合、肝毒性等六個模型。TOPKAT提供了化合物毒理性質的預測,對於有機化合物包含急性毒性、慢性毒性、誘變性、再生性等性質預測,同時也考慮環境毒性之預測。其QMRF格式為歐洲委員會聯合研究中心批准之正規文件。
如有任何問題,歡迎聯繫訊聯基因數位股份有限公司(原名:創源生技)
地址:台北市114內湖區新湖一路36巷28號
電話: (02) 2795 1777
網站: www.gga.asia

