

脊髓性肌肉萎縮症篩檢(SMA)

什麼是SMA脊髓性肌肉萎縮症?
脊髓性肌肉萎縮症(Spinal muscular atrophy, SMA)是一種可以致命的遺傳疾病,發病年齡從出生到成年皆有可能發生。當發病時,患者的肌肉會產生對稱性、逐漸性地退化且軟弱無力的萎縮表現,逐漸影響患者控制隨意肌肉的能力,如走路、爬行、吞嚥、呼吸和控制頭、頸肌肉等日常動作。
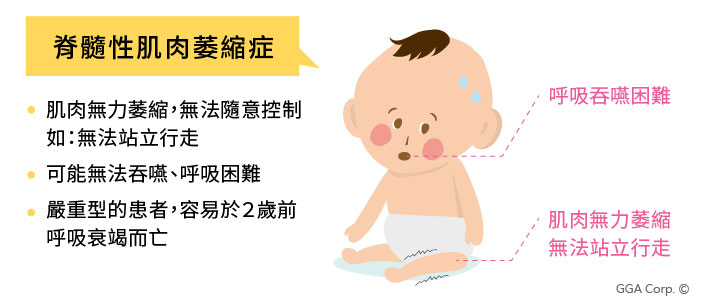
SMA脊髓性肌肉萎縮症明顯症狀
一般來說,脊髓性肌肉萎縮症依其發病年齡、疾病嚴重度及肌肉受影響程度,分為三型,第一型患者可能在嬰幼兒時期,就因呼吸衰竭而死亡,而第三型患者可能症狀較輕,壽命與一般人無異。
|
嚴重型 |
第一型脊髓性肌肉萎縮症發病年齡:約出生6個月內 |
|
中間型 |
第二型脊髓性肌肉萎縮症發病年齡:約出生後6-18個月間 |
|
輕型 |
第三型脊髓性肌肉萎縮症發病年齡:從一歲半至成年皆可能發生 |
SMA脊髓性肌肉萎縮治療藥物
-
以往此遺傳疾病僅能物理治療,改善患者四肢行動的靈活度、呼吸系統的照護,後來治療藥物Spinraza® (Nusinersen)經美國FDA核准上市,臨床結果顯示,在發病前就給予藥物的預防性治療,是最佳的黃金治療期,然而治療費用一次就需200多萬美元【資料來源1】,並非一般家庭可以負擔的費用。
-
儘管健保2020年已將SMA的治療藥物納入健保給付範圍,補助條件仍十分嚴苛。像是終身只需注射一次,即可達成治癒效果,一劑要價4,900萬新台幣的SMA用藥” Zolgensma®( -Onasemnogene abeparvovec-xioi)”,雖於2023年8月納入健保,但因價格高昂,為避免昂貴藥價排擠到其他罕藥納入給付的機會,健保署首次採「分期付款」的方式支付,給附條件也提高至「出生六個月內發病,且須經標準基因檢測方法確診」的患者,並且須經專家小組以特殊專案審查核准後使用,並非所有SMA病友都能獲得有效治療。
因此,在SMA帶因率也高的情況下,若能於懷孕前或孕期提早進行SMA脊髓性肌肉萎縮症帶因篩檢,確認是否有潛在的遺傳疾病風險,就能降低生下罕見兒的機率。
| 藥物名稱 | Spinraza 脊瑞拉 |
Zolgensma 諾健生 |
Evrysdi 服脊立 |
|---|---|---|---|
| 用藥方式 | 從腰椎穿刺於脊髓腔內注射 (基因剪輯) |
靜脈注射 (重組DNA) |
口服用粉劑 (基因剪輯) |
| 用藥次數 | 首年6劑 次年每年3劑 |
一次性 | 每日服用1次 |
| 納入健保時間 | 2020年7月 | 2023年8月 | 2020年 |
| 給付範圍 | 原本:限一歲前發病,首次治療未滿7歲者 擴增: 1. 3歲內發病確診 2. 若首次治療年滿7歲,則臨床評估運動功能指 標RULM≧15者 |
1. 出生六個月內發病 2. 經標準基因檢測確認為SMA患者 3. 經專家小組已特殊專案審查核准 |
原本:治療2歲以下病人 擴增:2個月以上,未滿18歲病患 |
| 藥價(台幣) | 約2百萬元/針 (10年療程約1億1355萬元) |
約4,900萬元/針 | 約918萬元/年 (療程約18年) |
資料來源:
1. https://www.biospace.com/article/phase-iii-trial-reinforces-value-of-novartis-gene-therapy-for-sma-/
2. SMA治療藥物爭取協會、衛生福利部中央健康保險署之全民健康保險藥物給付項目及支付標準、媒體報導整理


SMA脊髓性肌肉萎縮發生原因
目前所了解的脊髓性肌肉萎縮症,它的發生主要是因為基因產生突變所致。它的基因是位於第五條染色體長臂的區域,是一種稱為「運動神經元存活基因」(SMN1),約95%的脊髓肌肉萎縮症患者是因為SMN1的這段基因出現大片段缺失或轉換導致的,其它少數若無SMN1基因大片段缺失或轉換的患者,則可能是在SMN1基因上發生一些小突變而致病。大部份正常人具有二個以上之SMN1基因,帶因者只具有一個SMN1基因,而病人則完全沒有正常的 SMN1基因。
SMA脊髓性肌肉萎縮遺傳模式
-
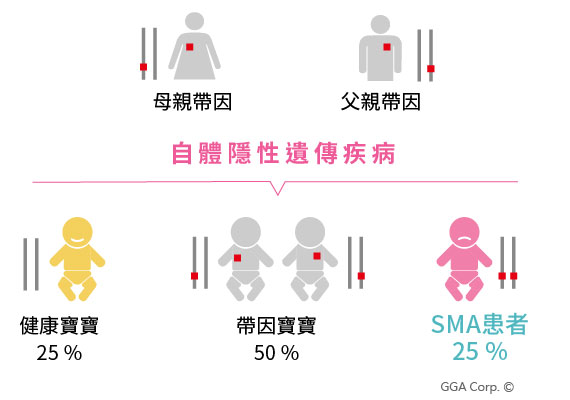
脊髓性肌肉萎縮症為體染色體隱性遺傳疾病,在台灣,這個疾病帶因率僅次於海洋性貧血,第二常見的遺傳疾病。
-
此病的帶因率非常高,約為1/40~1/60【資料來源1】,等於每40到60個人之中,就有一個是帶因者。若父母同為帶因者,則每一胎不管是男孩或女孩,都會有25%的機率成為脊髓性肌肉萎縮症的患者。以台灣每年有十幾萬名新生兒來算,一年就會新增十多名的脊髓性肌肉萎縮症病例【資料來源2】。
-
帶因是指一對染色體中其中一條染色體上帶有與隱性遺傳疾病相關的基因突變,且因為只有一條染色體有基因變異,另一條正常,所以帶因者通常健康與外觀上都無異常,許多人容易在不知情下,與同樣帶有相同疾病基因的另一半結婚,所以容易導致爸媽健康,卻生下患病小孩的情形。
資料來源:
1. 美國遺傳學會
2. 國民健康署
小心漏網之魚!SMA產前篩檢 恐存在5%的偽陰性
在高雄醫學大學附設醫院的門診中,曾有對夫妻於懷孕時接受SMA脊髓性肌肉萎縮症的帶因篩檢,報告顯示只有一人是典型的SMA帶因者,所以胎兒不會有SMA的患病問題,媽媽便繼續放心懷孕。沒想到等小孩生下來,新生兒篩檢竟發現孩子罹患了SMA脊髓性肌肉萎縮症!後續經遺傳諮詢建議,讓這位媽媽額外進行空間排列錯置的篩檢,才發現媽媽竟是少數的沈默帶因者(silent carrier),才會導致SMA帶因篩檢沒有抓到這類少數、但目前已可預防的風險!
市場上常規的SMA脊髓性肌肉萎縮症的帶因篩檢,多為檢測SMN1基因的數量,但少部分人可能出現SMN1基因的數量正常,但空間排列錯置的問題,造成SMA帶因篩檢難以分辨,這群人就被稱為「沉默帶因者」。儘管常規的SMA帶因篩檢技術已可排除約95%的帶因者,但這群SMN1基因數量與正常人無異的沉默帶因者,容易因無法被檢出,如同高醫案例中的夫妻,呈現報告偽陰性的現象。
訊聯基因(原名:創源)為提升脊髓性肌肉萎縮症帶因者的檢出率,特地從海外引進可針對沉默帶因者的篩檢技術,期望藉由SMN1基因的空間排列錯置風險評估,有效降低產前帶因篩檢的漏網之魚!(延伸閱讀:沉默帶因者的遺傳模式)
哪些人需做要SMA脊髓性肌肉萎縮篩檢
由於SMA脊髓性肌肉萎縮症的治療費用一般家庭難以負擔,除了以下列舉需要進行帶因篩檢外,美國婦產科醫學會建議孕婦在產前應接受SMA脊髓性肌肉萎縮症帶因篩檢,美國密蘇里州更通過法案,每位新生兒都需接受SMA脊髓性肌肉萎縮症篩檢。【資料來源1】
-
有SMA脊髓性肌肉萎縮症家族史的人
-
疑似有SMA脊髓性肌肉萎縮症帶因者
-
備孕、懷孕中的人
資料來源:
1.http://www.cureSMA.org
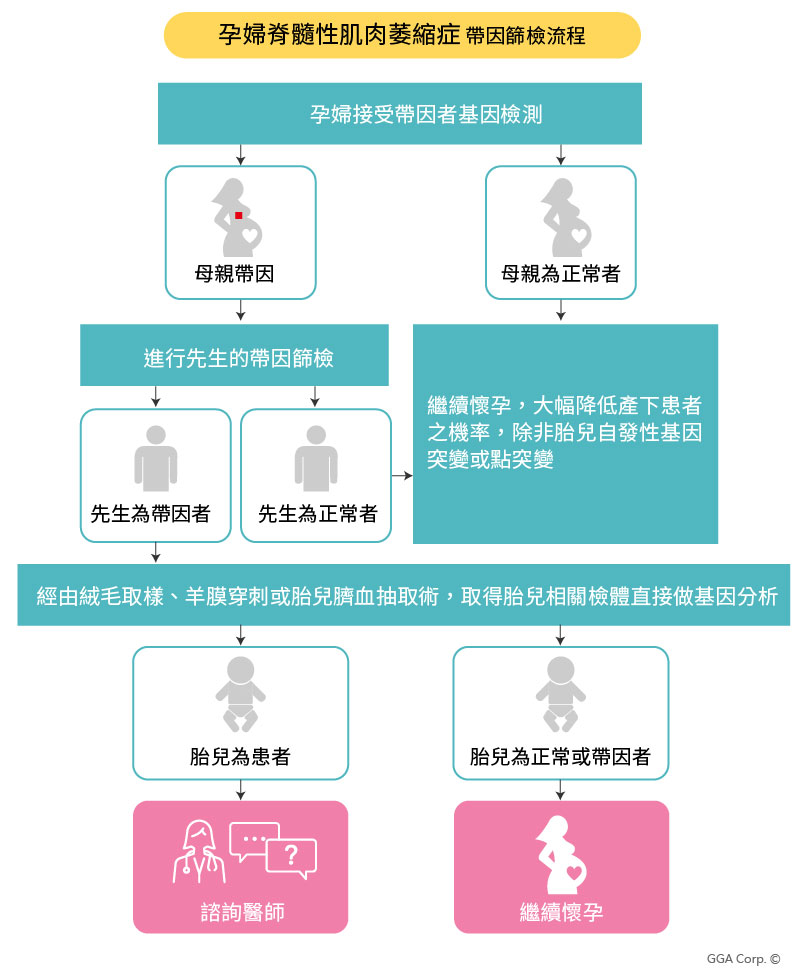
SMA脊髓性肌肉萎縮篩檢流程
-
於訊聯基因的合作醫院診所進行衛教諮詢
-
填寫檢測同意書
-
抽取血液進行SMA脊髓性肌肉萎縮症基因檢測
-
回院所觀看檢測報告
-
若孕婦為SMA帶因者,將請另一半進行SMA帶因篩檢
-
如果另一半同樣為SMA帶因者,孕婦將經由絨毛膜取樣、羊膜穿刺術或胎兒臍血抽取術,取得胎兒相關檢體直接做基因分析
-
如果胎兒為正常或是帶因者,孕婦可繼續懷孕;反之,若胎兒為SMA的患者,需諮詢醫師後續照護
為什麼SMA脊髓性肌肉萎縮篩檢選擇訊聯基因
-
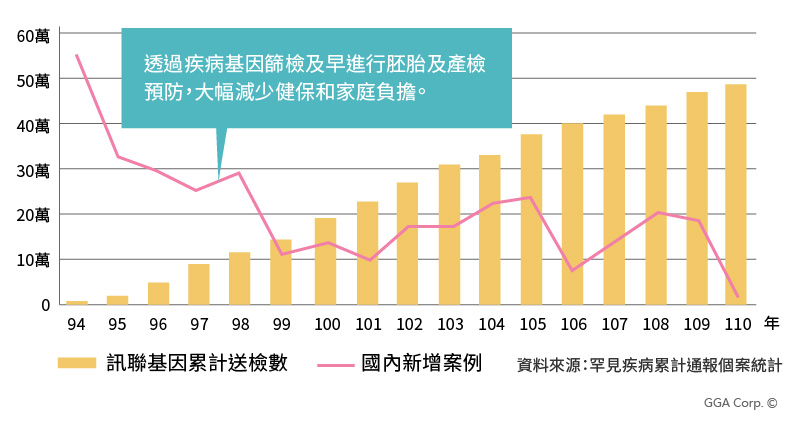
訊聯基因為全臺首家推出SMA脊髓性肌肉萎縮症帶因篩檢之生技公司,至今已有逾50萬人次接受創源SMA檢測,國健署統計的疾病通報案例逐年下降,為全臺灣每年省下40億醫藥費支出。
-
訊聯基因獨家引進SMN1基因的空間排列錯置風險評估,提高對SMA沉默帶因者的檢出機率。
-
由持有美國、臺灣兩地執照的資深遺傳諮詢師領軍的遺傳諮詢團隊,協助醫護人員進行遺傳諮詢評估,給予最安心的專業服務。
-
訊聯基因擁有國際認證的實驗室,且操作經驗超過20年,同時以專業團隊配送檢體,嚴格掌控檢體的品質。
-
訊聯基因擁有一條龍的檢測服務,若您夫妻雙方都有相同帶因,且有生育的需求,我們可銜接UPGD超快速胚胎著床前檢測服務,且擁有全台最大的臍帶血資料庫比對。
-
父母雙帶因時,訊聯基因補助當胎次SMA確診費用。
SMA脊髓性肌肉萎縮症常見問題
Q:為什麼要做脊髓性肌肉萎縮症(SMA)帶因篩檢?
A:此遺傳疾病目前尚無具體之治療方式可以治癒患者的症狀,僅能藉由物理治療改善患者四肢行動的靈活度、呼吸系統的照護,嚴重者需要依賴積極的支持性呼吸治療甚至仰賴呼吸器,這樣一來會造成家庭及社會很重的負擔。所以唯有依賴正確的帶因者篩檢流程,才能降此病的發生率。
Q:哪些人需要接受脊髓性肌肉萎縮症(SMA)帶因檢測呢?
A:由於帶因率相當高且帶因者沒有任何症狀,且目前此項檢驗相當簡單,只需如同一般抽血即可,因此,建議一般的民眾若打算生育或已經懷孕,最好都應接受SMA基因檢測。
Q:我和我先生都很正常,也沒有家族病史,有可能生下脊髓性肌肉萎縮症的小孩嗎?
A:全世界不論人種的差異,此病的帶原率約為1/40~1/60。過去由於缺乏正確快速的SMA帶因者診斷方式,一般沒有發病的民眾是無法得知自己是否帶有這樣的突變基因,而當兩個沒有任何症狀、且一切正常的帶因者結婚之後,每一次懷孕不論胎兒是男是女,都有25%的機會成為患者。我們建議每一對夫婦於結婚或懷孕後,先接受疾病帶因者的基因篩檢,若雙方皆為帶因者,建議於妊娠20周之前,接受產前胎兒基因診斷,以早期偵測。
Q:既然每一胎只有1/4的機會成為患者,那麼如果不幸生下一位SMA寶寶了,下一胎還會再懷有SMA寶寶嗎?
A:曾經生下脊髓性肌肉萎縮症患者,則表示父母雙方幾乎皆為帶因者(自發性突變也有,但非常少)。我們會強烈建議這類家庭的夫妻雙方,於之後每次懷孕一定要接受產前胎兒基因診斷,因為每一胎都會有1/4的機會生下這種小孩,此機會不會因前一胎不幸已生下一位此病之寶寶而有任何之減少,所以每一胎的產前基因診斷是絕對不可偷懶的。
Q:健保有給付脊髓性肌肉萎縮症(SMA)的治療嗎?
A:健保2020年已將SMA的治療藥物納入健保給付範圍,但補助條件十分嚴苛。像是2023年8月納入健保,一劑要價4,900萬新台幣的SMA用藥” Zolgensma®( -Onasemnogene abeparvovec-xioi)”,雖然終身只需注射一次,即可達成治癒效果,但因價格高昂,為避免昂貴藥價排擠到其他罕藥納入給付的機會,健保署首次採「分期付款」的方式支付,給附條件也提高至「出生六個月內發病,且須經標準基因檢測方法確診」的患者,並且須經專家小組以特殊專案審查核准後使用,並非所有SMA病友都能獲得有效治療。
因此,在SMA帶因率也高的情況下,若能於懷孕前或孕期提早進行SMA脊髓性肌肉萎縮症帶因篩檢,確認是否有潛在的遺傳疾病風險,就能降低生下罕見兒的機率。





